Study finds genetic contribution to human lifespan is about 50% – more than double previous estimates
Photo by Matteo Vistocco on Unsplash
What determines how long we live – and to what extent is our lifespan shaped by our genes? Surprisingly, scientists believed for decades that the heritability of human lifespan was relatively low compared to other human traits, standing at just 20 to 25%; some recent large-scale studies even placed it below 10%. Now, a new study from the Weizmann Institute of Science, published in Science, presents an entirely different picture. According to the findings, genetics accounts for about 50% of variation in human lifespan – twice as much, or more, than previously thought.
The study was led by Ben Shenhar from the lab of Prof Uri Alon of Weizmann’s Molecular Cell Biology Department.
“For many years, lifespan was attributed mainly to non-genetic factors, fuelling scepticism about genetic determinants of longevity”
Using mathematical models and analyses of three large twin databases from Sweden and Denmark – including, for the first time in this context, a dataset of twins who were raised apart – the researchers showed that earlier heritability estimates were masked by high levels of extrinsic mortality, such as deaths caused by accidents, infections and environmental hazards. Filtering out such extrinsic factors was impossible in historic datasets because they provided no information about the cause of death. To compensate for this limitation, the researchers developed an innovative framework that included mathematical simulation of virtual twins to separate deaths due to biological ageing from those caused by extrinsic factors. The new results are consistent with the heritability of other complex human traits and with findings from animal models.
Science Numbers
Up to age 80, the risk of dying from dementia shows a heritability of about 70% – far higher than that of cancer or heart disease.
The results have far-reaching implications for ageing research and public health. “For many years, human lifespan was thought to be shaped almost entirely by non-genetic factors, which led to considerable scepticism about the role of genetics in ageing and about the feasibility of identifying genetic determinants of longevity,” says Shenhar. “By contrast, if heritability is high, as we have shown, this creates an incentive to search for gene variants that extend lifespan, in order to understand the biology of ageing and, potentially, to address it therapeutically.”
An analysis of genetic data over nearly one million individuals shows that certain stretches of DNA, made up of short sequences repeated over and over, become longer and more unstable as we age. The study found that common genetic variants can speed up or slow down this process by up to fourfold, and that certain expanded sequences are linked to serious diseases including kidney failure and liver disease.
Why it matters
More than 60 inherited disorders are caused by expanded DNA repeats: repetitive genetic sequences that grow longer over time. These include devastating conditions like Huntington’s disease, myotonic dystrophy, and certain forms of ALS. Most people carry DNA repeats that gradually expand throughout their lives, but this instability and what genetic factors control it hadn’t been fully analysed within large biobanks. This study demonstrates that DNA repeat expansion is far more widespread than previously recognised and identifies dozens of genes that regulate this process, opening new avenues for developing treatments that could slow disease progression.
What the study did
Researchers from UCLA, the Broad Institute, and Harvard Medical School analysed whole-genome sequencing data from 490 416 UK Biobank participants and 414 830 All of Us Research Program participants. They developed new computational methods to detect and measure DNA repeat lengths and instability from standard sequencing data. The team examined 356 131 polymorphic repeat locations across the genome, tracking how repeat lengths changed with age in blood cells and identifying genetic variants that influenced expansion rates. They also searched for links between repeat expansions and thousands of disease outcomes to discover previously unknown disease associations.
What they found
Common DNA repeats in blood cells expand as people age. The researchers identified 29 genetic locations where inherited variants modified DNA repeat expansion rates, with effects varying up to fourfold between individuals with the highest and lowest genetic risk scores. Interestingly, the same DNA repair genes had opposite effects on different repeats: variants that stabilised some repeats destabilised others. The study also discovered that expansions in the GLS gene, which have a prevalence of around 0.03%, were associated with 14-fold higher risk of severe kidney disease and 3-fold higher risk of liver diseases, representing a newly recognised repeat expansion disorder.
What’s next
The findings establish blood-based DNA repeat measurements as potential biomarkers for testing future therapies aimed at slowing repeat expansion in diseases like Huntington’s. The research team’s computational tools can now be applied to other large biobank datasets to discover additional unstable repeats and disease associations. Understanding why the same genetic modifiers have opposite effects on different repeats will require detailed mechanistic studies of how DNA repair processes vary across cell types and genetic contexts. The discovery of GLS repeat-associated kidney and liver disease suggests additional unrecognised repeat expansion disorders may be lurking in biobank data, waiting to be found.
From the experts
“We found that most human genomes contain repeat elements that expand as we age,” said Margaux L. A. Hujoel, PhD, lead author of the study and assistant professor in the Departments of Human Genetics and Computational Medicine at the David Geffen School of Medicine at UCLA. “The strong genetic control of this expansion, with some individuals’ repeats expanding four times faster than others, points to opportunities for therapeutic intervention. These naturally occurring genetic modifiers show us which molecular pathways could be targeted to slow repeat expansion in disease.”
A Scottish patient has become the first person in the world to receive a pioneering therapy aimed at improving outcomes for those having heart bypass surgery. The treatment involves precisely editing DNA in veins to be used during heart bypass surgery to boost the production of a protective protein.
The treatment could help extend the lifespan of blood vessels used during the surgery and significantly improve patient health, experts say.
Cause of failures in bypass surgery
Heart bypass surgery – an operation to improve blood flow to the heart – is a life-saving treatment for patients with coronary heart disease.
The process typically uses one artery and two or more veins as bypass grafts – healthy blood vessels used to bypass a narrowed or blocked artery – creating a new route for blood to flow.
Vein grafts used in this type of surgery can fail because they are not naturally designed to withstand the high pressure of blood flow from the heart.
Protecting vein grafts
The PROTECT study, led by NHS Greater Glasgow and Clyde and the University of Glasgow in collaboration with NHS Golden Jubilee and the University of Edinburgh, is trialling a new gene therapy designed to support newly grafted blood vessels.
The treatment will introduce a gene, which produces a protein called TIMP-3, into the vein to be grafted.
TIMP-3 is involved in tissue remodelling. Higher levels of the protein could help to prevent thickening and blockage of the blood vessel over time, scientists say.
Exciting milestone
The research team has developed a way to treat the graft directly at the time of surgery, safely and efficiently delivering the gene therapy to the affected tissue before grafting into the heart.
It is hoped the treatment will help to extend a patient’s healthy life expectancy and reduce the need for further surgeries, experts say.
In a groundbreaking study published in the Proceedings of the National Academy of Sciences (PNAS), scientists at Pacific Northwest Research Institute (PNRI) have overturned a long-held belief in genetics: that inheriting two harmful variants in the same gene always worsens disease. Instead, the team found that, in many cases, two harmful variants can actually restore normal protein function.
The research focused on a human enzyme called argininosuccinate lyase (ASL), which plays a critical role in removing toxic ammonia from the body. Variants in ASL that decrease its activity cause one of the urea cycle disorders, a set of rare and potentially life-threatening metabolic diseases.
By experimentally measuring the functional impact of several thousand individual variants and variant combinations, PNRI researchers discovered that over 60% of pairs that were individually damaging could, together, bring enzyme activity back to healthy levels.
“This work shows that genetic variants don’t act independently in many important cases,” said Michelle Tang, PhD, PNRI Staff Scientist and lead author of the study. “For a defined group of genes, the default assumptions we use to predict disease risk simply don’t hold.”
This phenomenon is known as intragenic complementation. It occurs when damage caused by one variant is offset by another variant in a different part of the same protein. The mechanism was first proposed in 1964 by Francis Crick and Leslie Orgel, but until now had not been tested systematically or shown to be common or predictable at scale.
To make these interactions predictable, the research team developed an AI-based model that accurately forecasted whether two variants would restore protein function. The model achieved nearly 100% accuracy in predicting intragenic complementation in ASL as well as in a second human enzyme, fumarase, suggesting these rules apply broadly across the human genome.
“We’ve shown that, in many cases, two damaging variants can work together to restore protein function,“ said Aimée Dudley, PhD, PNRI Senior Investigator who led the study. “This kind of genetic interaction is not an isolated exception, but a widespread and underappreciated way that variants can interact, especially in rare disease contexts.”
The researchers estimate that approximately 4% of human genes have the structural features that allow this type of interaction. For these genes, standard genetic predictions can overestimate disease risk, particularly for people who carry two different variants in the same gene.
Cedars-Sinai scientists have developed an experimental drug that repairs DNA and serves as a prototype for a new class of medications that fix tissue damage caused by heart attack, inflammatory disease or other conditions.
“By probing the mechanisms of stem cell therapy, we discovered a way to heal the body without using stem cells,” said Eduardo Marbán, MD, PhD, executive director of the Smidt Heart Institute at Cedars-Sinai and the study’s senior author. “TY1 is the first exomer – a new class of drugs that address tissue damage in unexpected ways.”
TY1 is a laboratory-made version of an RNA molecule that naturally exists in the body. The research team was able to show that TY1 enhances the action of a gene called TREX1, which helps immune cells clear damaged DNA. In so doing, TY1 repairs damaged tissue.
The development of TY1 has been more than two decades in the making. It started when Marbán’s previous laboratory at Johns Hopkins University developed a technique to isolate progenitor cells from the human heart. Like stem cells, progenitor cells can turn into new healthy tissue, but in a more focused manner than stem cells. Heart progenitor cells promote the regeneration of the heart, for example.
Later, at Marbán’s lab at Cedars-Sinai, Ahmed Ibrahim, PhD, MPH, discovered that these heart progenitor cells send out tiny molecule-filled sacs called exosomes. These sacs are loaded with RNA molecules that help repair and regenerate injured tissue.
Ahmed Ibrahim, PhD, MPH“Exosomes are like envelopes with important information,” said Ibrahim, who is associate professor in the Department of Cardiology in the Smidt Heart Institute and first author of the paper. “We wanted to take apart these coded messages and figure out which molecules were, themselves, therapeutic.”
Scientists genetically sequenced the RNA material inside the exosomes. They found that one RNA molecule was more abundant than the others, hinting it might be involved in tissue healing. The investigators found the natural RNA molecule to be effective in promoting healing after heart attacks in laboratory animals. TY1 is the synthetic, engineered version of that RNA molecule, designed to mimic the structure of approved RNA drugs already in the clinic. TY1 works by increasing the production of immune cells that reverse DNA damage, a process that minimises the formation of scar tissue after a heart attack.
“By enhancing DNA repair, we can heal tissue damage that occurs during a heart attack,” Ibrahim said. “We are particularly excited because TY1 also works in other conditions, including autoimmune diseases that cause the body to mistakenly attack healthy tissue. This is an entirely new mechanism for tissue healing, opening up new options for a variety of disorders.”
The investigators next plan to study TY1 in clinical trials.
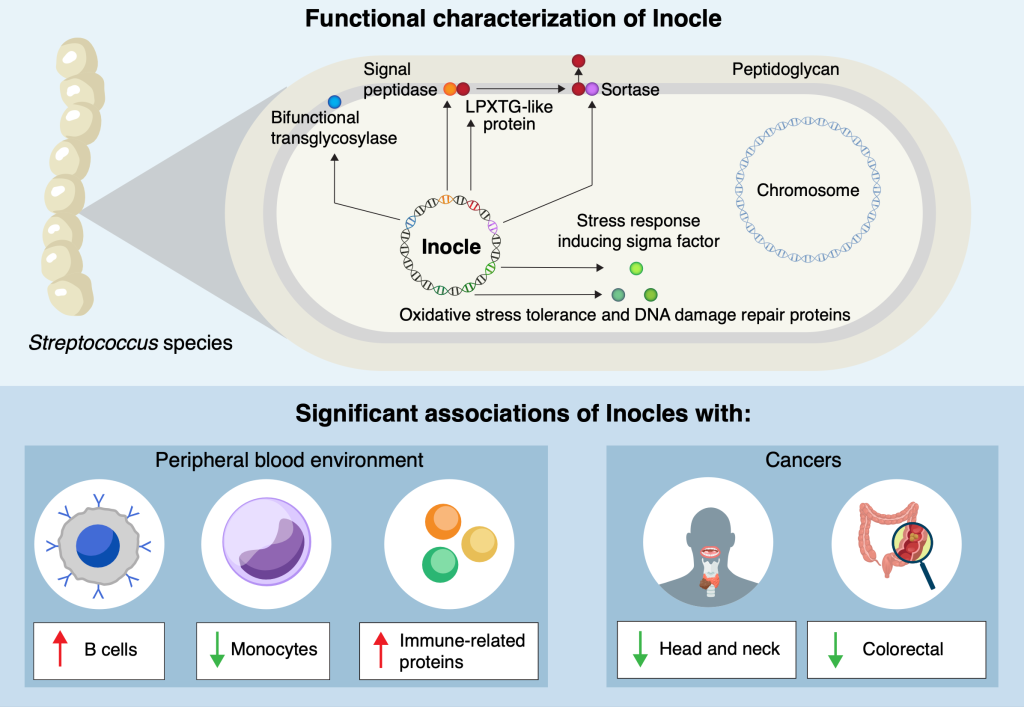
Researchers at the University of Tokyo and other institutions have made a surprising discovery hiding in people’s mouths: Inocles, giant DNA elements that had previously escaped detection. These appear to play a central role in helping bacteria adapt to the constantly changing environment of the mouth. The findings provide fresh insight into how oral bacteria colonise and persist in humans, with potential implications for health, disease and microbiome research.
You might think that modern medical science knows everything there is to know about the human body. But even within the last decade, small, previously unknown organs have been discovered, and there’s one area of human biology that is currently going through a research renaissance, the microbiome. This includes familiar areas such as the gut microbiome, but also the oral microbiome. Inspired in part by recent discoveries of extraneous DNA in the microbiome of soil, Project Research Associate Yuya Kiguchi and his team turned their sights to a large set of saliva samples collected by the Yutaka Suzuki Lab of the Graduate School of Frontier Sciences at the University of Tokyo. They wondered if they might find something similar in human saliva.
“We know there are a lot of different kinds of bacteria in the oral microbiome, but many of their functions and means of carrying out those functions are still unknown,” said Kiguchi. “By exploring this, we discovered Inocles, an example of extrachromosomal DNA – chunks of DNA that exist in cells, in this case bacteria, but outside their main DNA. It’s like finding a book with extra footnotes stapled to it, and we’re just starting to read them to find out what they do.”
Detecting Inocles was not easy, as conventional sequencing methods fragment genetic data, making it impossible to reconstruct large elements. To overcome this, the team applied advanced long-read sequencing techniques, which can capture much longer stretches of DNA. A key breakthrough came from co-first author Nagisa Hamamoto, who developed a method called preNuc to selectively remove human DNA from saliva samples, greatly improving the quality of sequencing long sections of other DNA. This allowed the researchers to assemble for the first time complete Inocle genomes, which turned out were hosted by the bacteria Streptococcus salivarius, though identifying the host itself was a difficult matter.
“The average genome size of Inocle is 350 kilobase pairs, a measure of length for genetic sequences, so it is one of the largest extrachromosomal genetic elements in the human microbiome. Plasmids, other forms of extrachromosomal DNA, are at most a few tens of kilobase pairs,” said Kiguchi. “This long length endows Inocles with genes for various functions, including resistance to oxidative stress, DNA damage repair and cell wall-related genes, possibly involved in adapting to extracellular stress response.”
The team aims to develop stable methods for culturing Inocle-containing bacteria. This will allow them to investigate how Inocles function, whether they can spread between individuals, and how they might influence oral health conditions such as cavities and gum disease. Since many Inocle genes remain uncharacterised, researchers will use a mixture of laboratory experiments and also computational simulations such as AlphaFold to predict and model the roles Inocles may play.
“What’s remarkable is that, given the range of the human population the saliva samples represent, we think 74% of all human beings may possess Inocles. And even though the oral microbiome has long been studied, Inocles remained hidden all this time because of technological limitations,” said Kiguchi. “Now that we know they exist, we can begin to explore how they shape the relationship between humans, their resident microbes and our oral health. And there’s even some hints that Inocles might serve as markers for serious diseases like cancer.”
University of Hawaiʻi at Mānoa scientists have uncovered a direct link between a missing Y chromosome gene and male infertility. Their new research reveals that deleting this single gene in mice not only caused infertility but also disrupted hundreds of other genes vital for healthy sperm. The findings, published August 27 in Cell Death and Differentiation, offer significant implications for understanding reproductive health.
Using CRISPR gene-editing, the team created mice missing one or both versions. Males without both, known as Zfy double knockouts, were completely infertile, with severely abnormal or absent sperm.
“This work really pushes forward our understanding of how this important Zfy gene works,” said Ward. “We identified pathways and other genes that are affected and we can now study how exactly Zfy regulates them.”
To continue investigations, the researchers turned to assisted reproduction techniques pioneered at UH, including intracytoplasmic sperm injection (ICSI) and round spermatid injection (ROSI). This allowed them to examine the molecular consequences of Zfy loss.
When one gene disrupts hundreds
The results revealed that without Zfy, hundreds of genes became misregulated – some too active, others too weak. Many of these genes are responsible for sperm production, DNA packaging, and cell survival.
As a result, sperm precursor cells in the testes died off early, and the sperm that did form carried fragile DNA that wasn’t properly condensed.
The study details can be found in an article published in Cell Death and Differentiation, a leading peer-reviewed journal.
Couple or siblings? New study may explain why we prefer partners who are similar to us. Photo by Daniil Onischenko on Unsplash
It is no secret that people are often drawn to romantic partners who seem similar to themselves. This tendency, called assortative mating, has been established in humans (Horwitz et al., 2023; Luo, 2017) as well as other species. Fish, for example, demonstrate the behaviour frequently (Jiang et al., 2013).
Assortative mating has also recently been in focus on social media with the viral Siblings or Dating game, where people guess whether two individuals who look alike are related or a couple.
The idea is well-founded in academic research. Humans have been observed to select partners with similar physical, personality, and demographic traits (Horwitz et al., 2023), which can impact the genetics of populations – creating subgroups that emphasise the presence of shared traits (Abdellaoui et al., 2015).
But selecting a partner like ourselves may not be solely determined by personal choice. A new study soon to be published in Psychological Science suggests that assortative mating can be explained relatively simply by looking at the inheritance of preferred traits and corresponding preferences for those traits.
Coauthors Kaitlyn Harper and Brendan Zietsch from the University of Queensland describe this scenario simply: If you are tall, you may have inherited tallness from one parent (say, your mother) and the preference for tallness in a romantic partner from your other parent (in this case, your father). The combination of those inherited traits means that you exist in the world as a tall person and are attracted to tall people.
The idea that preference for a particular trait could lead to genetic correlations has been discussed in previous research but is a newer concept for evolutionary psychology, especially in the context of assortative mating.
“The pieces were there, but they hadn’t been connected in this way before,” Harper said. “Agent-based modelling helped us connect the dots – by simulating populations, we could see that assortative mating naturally emerged without the need for additional assumptions or processes.”
She added that this research wouldn’t have been possible without an interdisciplinary mindset.
“The mechanism itself is familiar in evolutionary biology, but it wasn’t thought of as an explanation for assortative mating,” she said. “Making that connection only became possible when we looked across the two disciplines.”
To test this theory, the authors ran an agent-based model where partners are chosen according to heritable traits and preferences over 100 generations. They included models with and without selection pressure on the number of offspring within each generation to assess how the theory stands up under more naturalistic conditions.
They found that even with up to 10 preferences for traits in a partner, clear genetic correlations formed between traits and preferences for those traits, which resulted in the agents choosing partners similar to themselves. Models with selection pressure generated less-stable correlations, which the authors attribute to reduced variance in traits.
“The power of this finding is in its parsimony – it shows that a phenomenon which has puzzled researchers for decades can be understood through an explanation that was hiding in plain sight,” Harper said. “And because the mechanism is so general, it can also apply to assortative mating in animals, where many of the explanations proposed for humans wouldn’t make sense.”
Animals that hibernate are incredibly resilient. They can spend months without food or water, muscles refusing to atrophy, body temperature dropping to near freezing as their metabolism and brain activity slow to a crawl. When they emerge from hibernation, they recover from dangerous health changes similar to those seen in type 2 diabetes, Alzheimer’s disease, and stroke.
New genetic research suggests that hibernating animals’ superpowers could lie hidden in human DNA – with clues on how to unlock them, perhaps one day leading to treatments that could reverse neurodegeneration and diabetes.
A gene cluster called the “fat mass and obesity (FTO) locus” plays an important role in hibernators’ abilities, the researchers found. Intriguingly, humans have these genes too. “What’s striking about this region is that it is the strongest genetic risk factor for human obesity,” says Chris Gregg, PhD, professor in neurobiology and human genetics at University of Utah Health and senior author on the studies. But hibernators seem able to use genes in the FTO locus in new ways to their advantage.
The team identified hibernator-specific DNA regions that are near the FTO locus and that regulate the activity of neighbouring genes, tuning them up or down. The researchers speculate that adjusting the activity of neighbouring genes, including those in or near the FTO locus, allows hibernators to pack on the pounds before settling in for the winter, then slowly use their fat reserves for energy throughout hibernation.
Indeed, the hibernator-specific regulatory regions outside of the FTO locus seem crucial for tweaking metabolism. When the researchers mutated those hibernator-specific regions in mice, they saw changes in the mice’s weight and metabolism. Some mutations sped up or slowed down weight gain under specific dietary conditions; others affected the ability to recover body temperature after a hibernation-like state or tuned overall metabolic rate up or down.
Intriguingly, the hibernator-specific DNA regions the researchers identified weren’t genes themselves. Instead, the regions were DNA sequences that contact nearby genes and turn their expression up or down, like an orchestra conductor fine-tuning the volume of many musicians. This means that mutating a single hibernator-specific region has wide-ranging effects extending far beyond the FTO locus, explains Susan Steinwand, research scientist in neurobiology at U of U Health and first author on one of the studies. “When you knock out one of these elements – this one tiny, seemingly insignificant DNA region – the activity of hundreds of genes changes,” she says. “It’s pretty amazing.”
Understanding hibernators’ metabolic flexibility could lead to better treatments for human metabolic disorders like type 2 diabetes, the researchers say. “If we could regulate our genes a bit more like hibernators, maybe we could overcome type 2 diabetes the same way that a hibernator returns from hibernation back to a normal metabolic state,” says Elliott Ferris, MS, bioinformatician at U of U Health and first author on the other study.
Uncovering the regulation of hibernation
Finding the genetic regions that may enable hibernation is a problem akin to excavating needles from a massive DNA haystack. To narrow down the regions involved, the researchers used multiple independent whole-genome technologies to ask which regions might be relevant for hibernation. Then, they started looking for overlap between the results from each technique.
First, they looked for sequences of DNA that most mammals share but that had recently changed in hibernators. “If a region doesn’t change much from species to species for over 100 million years but then changes rapidly and dramatically in two hibernating mammals, then we think it points us to something that is important for hibernation, specifically,” Ferris says.
To understand the biological processes that underlie hibernation, the researchers tested for and identified genes that turn up or down during fasting in mice, which triggers metabolic changes similar to hibernation. Next, they found the genes that act as central coordinators, or “hubs,” of these fasting-induced changes to gene activity.
Many of the DNA regions that had recently changed in hibernators also appeared to interact with these central coordinating hub genes. Because of this, the researchers expect that the evolution of hibernation requires specific changes to the controls of the hub genes. These controls comprise a shortlist of DNA elements that are avenues for future investigation.
Awakening human potential
Most of the hibernator-associated changes in the genome appeared to “break” the function of specific pieces of DNA, rather than confer a new function. This hints that hibernators may have lost constraints that would otherwise prevent extreme flexibility in the ability to control metabolism. In other words, it’s possible that the human “thermostat” is locked to a narrow range of continuous energy consumption. For hibernators, that lock may be gone.
Hibernators can reverse neurodegeneration, avoid muscle atrophy, stay healthy despite massive weight fluctuations, and show improved aging and longevity. The researchers think their findings show that humans may already have the needed genetic code to have similar hibernator-like superpowers—if we can bypass some of our metabolic switches.
“Humans already have the genetic framework,” Steinwand says. “We just need to identify the control switches for these hibernator traits.” By learning how, researchers could help confer similar resilience to humans.
Genetic analysis of the early pandemic virus shows key adaptations to humans.
Creative artwork featuring colourised 3D prints of influenza virus (surface glycoprotein hemagglutinin is blue and neuraminidase is orange; the viral membrane is a darker orange). Note: Not to scale. Credit: NIAID
Researchers from the universities of Basel and Zurich have used a historical specimen from UZH’s Medical Collection to decode the genome of the virus responsible for the 1918-1920 influenza pandemic in Switzerland. The genetic material of the virus reveals that it had already developed key adaptations to humans at the outset of what became the deadliest influenza pandemic in history.
New viral epidemics pose a major challenge to public health and society. Understanding how viruses evolve and learning from past pandemics are crucial for developing targeted countermeasures. The so-called Spanish flu of 1918-1920 was one of the most devastating pandemics in history, claiming some 20 to 100 million lives worldwide. And yet, until now, little has been known about how that influenza virus mutated and adapted over the course of the pandemic.
More than 100-year-old flu virus sequenced
An international research team led by Verena Schünemann, a paleogeneticist and professor of archaeological science at the University of Basel (formerly at the University of Zurich) has now reconstructed the first Swiss genome of the influenza virus responsible for the pandemic of 1918-1920. For their study, the researchers used a more than 100-year-old virus taken from a formalin-fixed wet specimen sample in the Medical Collection of the Institute of Evolutionary Medicine at UZH. The virus came from an 18-year-old patient from Zurich who had died during the first wave of the pandemic in Switzerland and underwent autopsy in July 1918.
Three key adaptations in Swiss virus genome
“This is the first time we’ve had access to an influenza genome from the 1918-1920 pandemic in Switzerland. It opens up new insights into the dynamics of how the virus adapted in Europe at the start of the pandemic,” says last author Verena Schünemann. By comparing the Swiss genome with the few influenza virus genomes previously published from Germany and North America, the researchers were able to show that the Swiss strain already carried three key adaptations to humans that would persist in the virus population until the end of the pandemic.
Two of these mutations made the virus more resistant to an antiviral component in the human immune system – an important barrier against the transmissions of avian-like flu viruses from animals to humans. The third mutation concerned a protein in the virus’s membrane that improved its ability to bind to receptors in human cells, making the virus more resilient and more infectious.
New genome-sequencing method
Unlike adenoviruses, which cause common colds and are made up of stable DNA, influenza viruses carry their genetic information in the form of RNA, which degrades much faster. “Ancient RNA is only preserved over long periods under very specific conditions. That’s why we developed a new method to improve our ability to recover ancient RNA fragments from such specimens,” says Christian Urban, the study’s first author from UZH. This new method can now be used to reconstruct further genomes of ancient RNA viruses and enables researchers to verify the authenticity of the recovered RNA fragments.
Invaluable archives
For their study, the researchers worked hand in hand with UZH’s Medical Collection and the Berlin Museum of Medical History of the Charité University Hospital. “Medical collections are an invaluable archive for reconstructing ancient RNA virus genomes. However, the potential of these specimens remains underused,” says Frank Rühli, co-author of the study and head of the Institute of Evolutionary Medicine at UZH.
The researchers believe the results of their study will prove particularly important when it comes to tackling future pandemics. “A better understanding of the dynamics of how viruses adapt to humans during a pandemic over a long period of time enables us to develop models for future pandemics,” Verena Schünemann says. “Thanks to our interdisciplinary approach that combines historico-epidemiological and genetic transmission patterns, we can establish an evidence-based foundation for calculations,” adds Kaspar Staub, co-author from UZH. This will require further reconstructions of virus genomes as well as in-depth analyses that include longer intervals.