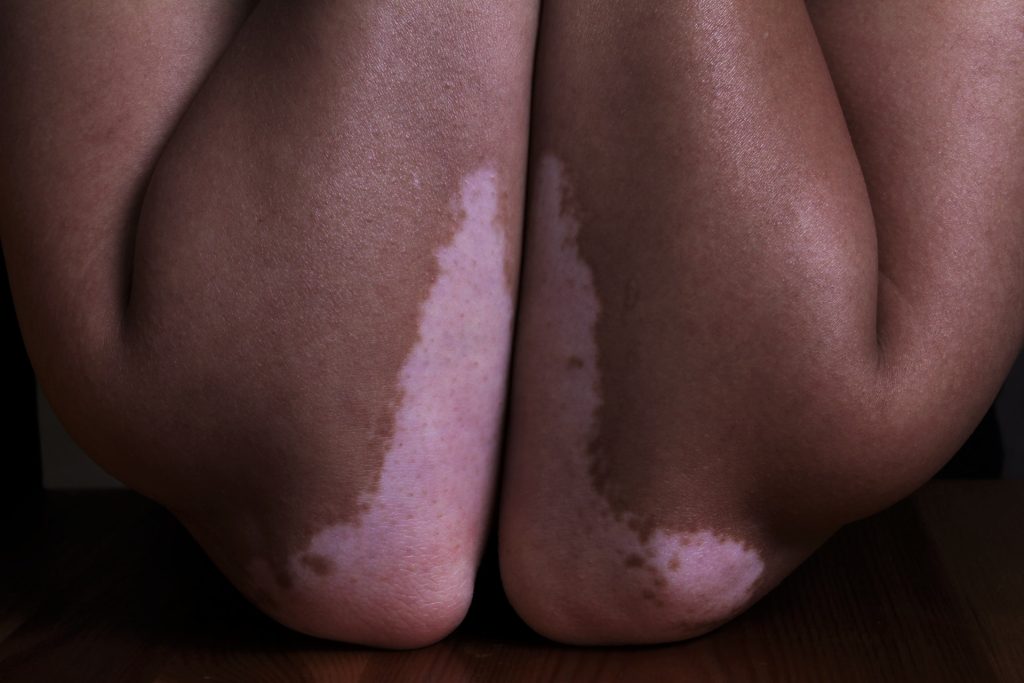
Targeting keratinocyte metabolism could be a new method of vitiligo treatment. Photo by Hanen BOUBAHRI on Unsplash
A new study published today in JCI Insight reveals the unique cell-to-cell communication networks that can perpetuate inflammation and prevent repigmentation in patients with stable vitiligo disease, and the particular role that keratinocytes play.
“In this study, we couple advanced imaging with transcriptomics and bioinformatics to discover the cell-to-cell communication networks between keratinocytes, immune cells and melanocytes that drive inflammation and prevent repigmentation caused by vitiligo,” said Anand K. Ganesan, MD, PhD, professor at University of California, Irvine. “This discovery will enable us to determine why white patches continue to persist in stable vitiligo disease, which could lead to new therapeutics to treat this disease.”
Vitiligo is an autoimmune skin disease characterised by the progressive destruction of melanocytes by immune cells called autoreactive CD8+ T cells, resulting in disfiguring patches of white depigmented skin. This disease has shown to cause significant psychological distress among patients. Melanocyte destruction in active vitiligo is mediated by CD8+ T cells, but until now, why the white patches in stable disease persist was poorly understood.
“Until now, the interaction between immune cells, melanocytes, and keratinocytes in situ in human skin has been difficult to study due to the lack of proper tools,” said Jessica Shiu, MD, PhD, assistant professor of dermatology and one of the first authors of the study. “By combining non-invasive multiphoton microscopy (MPM) imaging and single-cell RNA sequencing (scRNA-seq), we identified distinct subpopulations of keratinocytes in lesional skin of stable vitiligo patients along with the changes in cellular compositions in stable vitiligo skin that drive disease persistence. In patients that responded to punch grafting treatment, these changes were reversed, highlighting their role in disease persistence.”
MPM is a unique tool that has broad applications in human skin. MPM is a noninvasive imaging technique capable of providing images with sub-micron resolution and label-free molecular contrast which can be used to characterise keratinocyte metabolism in human skin.
Most studies on vitiligo have focused on active disease, while stable vitiligo remains somewhat of a mystery. Studies are currently investigating when metabolically altered keratinocytes first appear and how they may affect the repigmentation process in patients undergoing treatment.
The study findings suggest the possibility of targeting keratinocyte metabolism in vitiligo treatment. Further studies are needed to improve the understanding of how keratinocyte states affect the tissue microenvironment and contribute to disease pathogenesis.
A common culprit of skin and respiratory infections, Staphylococcus aureus is highly unpredictable, with the bacteria mostly harmlessly present in the skin of 20–30% of people. However, these bacteria can occasionally cause infections that lead to deadly complications, such as pneumonia, deep skin infections, and sepsis. This was a totally unpredictable outcome – until now.
Now, a new study published in Science identifies a mutated gene common to multiple patients who suffer life-threatening infections and suggests that people living with a genetic condition known as 5p- or Cri-du-chat syndrome may be at similar risk.
“We have characterised severe Staphylococcus aureus infection at the genetic, cellular, immunological, and clinical levels,” said András Spaan, the study’s first author. “By integrating these levels, we have established causality and provided clues for future interventions.”
A first for cell intrinsic immunity
To find out why S. aureus causes disease in some people but not others, scientists examined the protein-coding genomes of more than 100 patients who had suffered from unexplained severe staph infections.
The common genetic thread linking some of these disparate patients were mutations of a gene called OTULIN, which is perched along the short arm of chromosome 5 and codes for an enzyme involved in regulating inflammation. These individuals were not entirely bereft of OTULIN –only one of their two copies of the gene was mutated – but that deficiency appeared to be all it took to render them vulnerable to infections that would scarcely harm other people.
The scientists expected to find that OTULIN deficiency somehow cripples white blood cells or otherwise prevents the immune system from snuffing out S. aureus. But further investigation revealed that these mutations indirectly cause an unrelated protein to aggregate on the surfaces of skin and lung cells, gumming up the tools that those cells use to defend themselves from a toxin produced by S. aureus. This mechanism of defense is known as cell intrinsic immunity.
This finding was particularly surprising because, until then, specific defects in cell intrinsic immunity had only been linked to a predisposition to some viral infections, from COVID to herpes to encephalitis. It had never been shown to play a role in bacterial disease. “This is the first known instance of cell intrinsic immunodeficiency predisposing patients to bacterial infection,” Spaan says.
A larger role for OTULIN
While the individuals whom Spaan and colleagues studied were only missing one copy of OTULIN, people born without either functional copy of this gene face a bevy of early-onset inflammatory diseases, which often prove fatal in the first year of life.
This observation led Spaan to conclude that one functional copy of OTULIN is enough to prevent inflammatory disease, but insufficient to protect against life-threatening staph infections—a genetic mechanism known as haploinsufficiency. “The genetic mechanism was important to pin down,” Spaan says. “People with two functional copies of the gene appear to be healthy, those with no functional copies have autoinflammatory disease, and those with one functional copy are susceptible to severe staph infections.”
Given that general rule, the researchers hypothesized that any population missing only one copy of OTULIN would be similarly predisposed to severe infections. So they then examined a group of volunteers with 5p- syndrome, the most common chromosomal deletion disorder in humans characterized by developmental delays, intellectual disabilities and, in infants, a high-pitched cry. Most 5p- syndrome patients are missing the entire short arm of chromosome 5 and therefore invariably go about their lives with only one functional copy of OTULIN.
Indeed, upon examining six 5p- syndrome patients, the team found that one third were susceptible to lung infections. “We were able to demonstrate that this susceptibility is driven by the fact that they had only one functional copy of OTULIN,” Spaan says. “In many ways, these patients looked genetically similar to the patients we had identified with severe staph infections.”
“Both clinically, and on the cellular level, they could almost be said to have the same disease.”
The findings do not imply that everyone with OTULIN haploinsufficiency or 5p- syndrome will contract severe infections. In fact, the initial results of the study suggested that only 30 percent of individuals with these mutations develop severe disease. Why OTULIN haploinsufficiency appears to cause disease in some patients but not others—a common phenomenon that genetics researchers call “incomplete penetrance”—will be the subject of follow-up studies.
“Many genetic disorders act in this way, but it remains puzzling,” Spaan says. “Why are some people with these mutations perfectly healthy, while others get super ill and may even die?”
One potential answer has already surfaced. Spaan and colleagues found that individuals with OTULIN mutations but no sign of severe disease had high levels of antibodies that neutralise the toxin produced by S. aureus, perhaps due to prior exposure to the common skin bacteria. Individuals with severe disease, on the other hand, had precious few antibodies.
Further investigation into genetic predisposition to diseases, particularly ones as stubborn as staphylococcal infections, may help the development of future treatments. “Studies on these disorders can act as a compass,” Spaan said, “Our research clarifies the interactions between hosts and pathogens, revealing scientific insights into pathogenesis and immunity.”
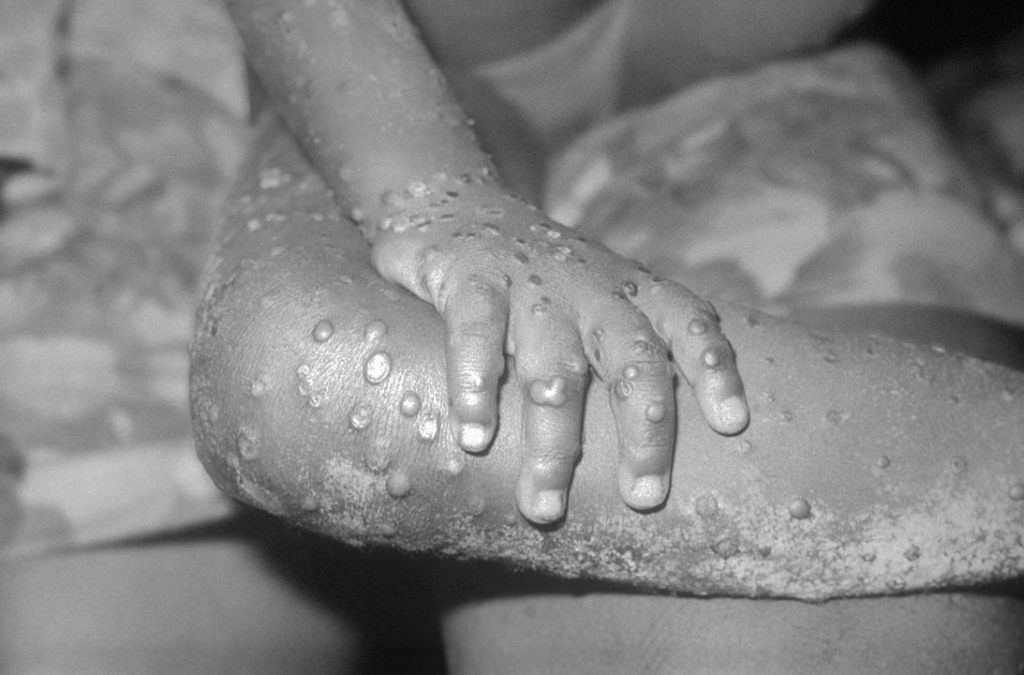
Close-up of monkeypox lesions on the arm and leg of a female child. Credit: Wikimedia Commons
On Friday, May 20, the World Health Organization has reported that there were 80 cases of monkeypox reported in 12 countries, but has not mentioned which countries those are. However, the National Institute for Communicable Diseases has not reported any cases in South Africa, though there has now been a case reported in Australia.
Update: as of 23 May, the NICD has reported that there are 145 cases in 15 countries, but confirms there are no local cases.
Normally endemic to certain countries where it resides in animal reservoirs, monkeypox is rarely encountered in countries outside those regions. The WHO notes that this is “atypical” for the zoonotic orthopoxvirus, which causes smallpox-like symptoms but with a lower mortality. European public health agencies have so far reported that the UK, Spain, Portugal, Germany, Belgium, France, the Netherlands, Italy and Sweden have seen cases. The first patient in the UK with the virus had returned from a trip to Nigeria, likely catching it there. Cases have been reported in the US and Canada.
The WHO advises that, “As monkeypox spreads through close contact, the response should focus on the people affected and their close contacts. People who closely interact with someone who is infectious are at greater risk for infection: this includes health workers, household members and sexual partners.”
At present, it is unclear why this unusual outbreak is happening now, especially amid the heightened vigilance of the COVID pandemic. One possibility is that some mutation is responsible, though there is little evidence at present to suggest a new variant is responsible.
Another explanation could be that this is simply a matter of the right place and time for the virus. It may also be easier for monkeypox to spread nowadays compared to when there was more widespread use of smallpox vaccine.
A large study has found that treatment with methylprednisolone – a cheap, widely used corticosteroid – halves the risk of losing kidney function and kidney failure in IgA nephropathy. The study, published in the journal JAMA, also found that this can be effectively achieved with fewer side effects if a reduced dose is used.
Researchers say the results of the multi-country study will provide a clear treatment option with definite benefits outweighing well defined and mostly manageable risks.
IgA nephropathy is a common form of glomerulonephritis caused by the deposition of IgA immunoglobulins in the glomerular basement membrane. Immune-mediated damage to the basement membrane results in haematuria and renal insufficiency progressing to kidney failure in some.
Joint Principal Investigator Professor Vlado Perkovic said that around 10–30% of people with the condition go on to develop kidney failure that requires dialysis or kidney transplantation to prevent death.
“There are few proven treatment options so many treatments including corticosteroids have been used in some patients for decades, despite uncertainty about their effectiveness, as well as the ideal dose. This has led to significant regional variability and clinical uncertainty about this treatment,” he said.
The Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) study is a double-blinded, randomised, controlled trial that assessed the effects of oral methylprednisolone on major kidney outcomes, kidney failure and safety in patients with IgA nephropathy.
503 patients diagnosed with IgA nephropathy were recruited from centres across Australia, Canada, China (including Hong Kong), India and Malaysia between May 2012 and November 2019. Patients were randomised to receive methylprednisolone or a placebo at:
full dose of 0.6-0.8mg/kg per day of methylprednisolone or placebo for 2 months reducing by 8mg per day each month (262 participants between May 2012 and November 2015), or
reduced dose of 0.4mg/kg per day of methylprednisolone or placebo, also for two months, reducing to 4mg per day each month (241 participants between March 2017 and November 2019),
for a total treatment period of 6–9 months.
“We found that that treatment with methylprednisolone for six to nine months significantly reduced the risk of losing substantial kidney function, kidney failure requiring dialysis or transplantation, or death from kidney disease compared to placebo,” said Professor Perkovic.
“However, there was an increase in serious adverse events in those who received methylprednisolone, mainly seen in the full dose regimen with fewer in the reduced dose treatment group.”
Joint Principal Investigator Professor Hong Zhang said that with IgA nephropathy being an immune-mediated condition, the benefits seen were likely due to the immune suppressing action of the steroid treatment.
“A well-known side effect of steroid treatment is an increased risk of infections, but we found that this could be mitigated to a degree by using the lower dose and giving the patients antibiotics to prevent infections,” she said.
“This is the strongest evidence yet for the benefit of any treatment for the prevention of kidney failure in people with IgA nephropathy.
“The results provide a treatment option for clinicians and patients, especially at the lower dose, given the net benefits versus the risk of side effects,” she added.
Associate Professor Muh Geot Wong said that given that the condition develops slowly, and that there was some indication that the effects of treatment appeared to diminish over time, the research team have now extended the study.
“We are now following a significant number of patients from our original study for another five years so we will have a total of around ten years follow up,” he said.
“By then, we hope to have the most comprehensive set of evidence ever collected to help guide the treatment of people with this type of kidney disease.”
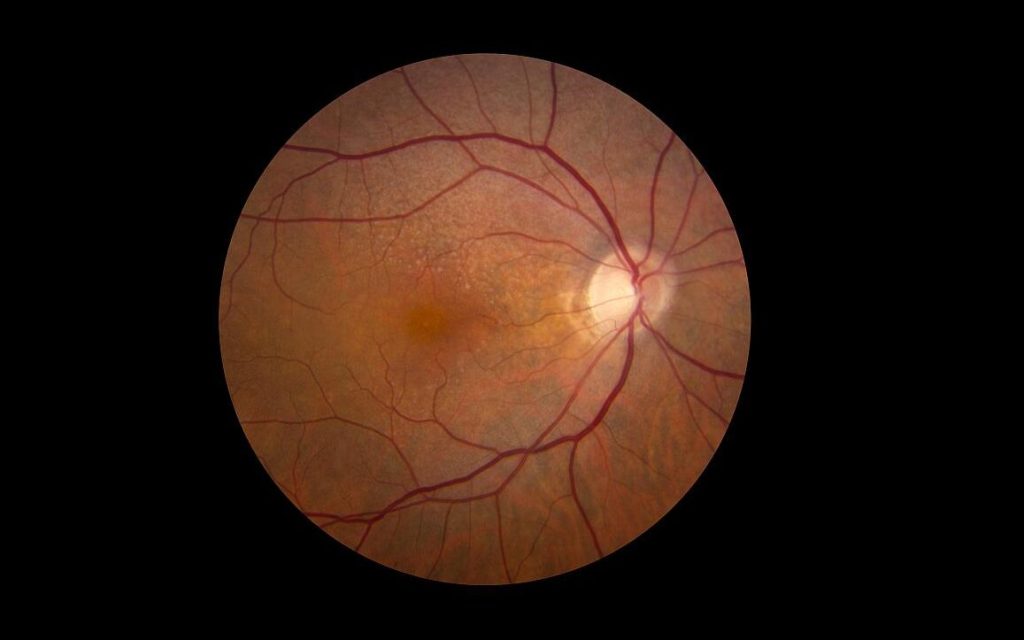
In an important step in treating a major cause of blindness, scientists have successfully identified early signs of age-related macular degeneration (AMD), in which higher number of mast cells are observed. This finding could be exploited by new treatments before symptoms develop. The study is published in PNAS.
Scientists have long known that people with certain genes on chromosomes 1 and 10 have a 2- to 3-fold higher risk of developing AMD, although lifestyle factors also play a role.
The team identified higher numbers of mast cells in the eyes of people when either of the risk genes were present, even when there were symptoms, suggesting an early mechanism in common.
They also showed the mast cells release enzymes in the back of the eye which then damage structures underneath the retina that in time is likely to damage the retina itself.
Mast cells exist in most tissues and are one of the immune system’s first defenses against infection, especially parasitic disease and damage.
Scientists already know there are more mast cells in the choroid in people with established AMD. The current study, however, identified higher levels in people before the disease develops.
The genes on chromosome 1 are linked to a part of the immune system called the complement cascade, which is associated with a risk of AMD.
Though the functional role of genes expressed by chromosome 10 are not known, but increased risk of AMD is.
Dr Richard Unwin, one of the study leaders, said: “What is really exciting about this work is that we are studying tissue from people before they have signs of the disease. This gives us a look into the very earliest stages, and gives us hope that we can intervene to stop the disease developing and ultimately prevent loss of vision”
The scientists used healthy human eye tissue donated post mortem to the Manchester Eye Tissue Repository.
They identified those who are at risk of developing age-related macular degeneration based on their risk genes, and discovered underlying changes in the tissue of the otherwise healthy at-risk individuals.
They collected retinal tissue from the back of donor eyes post mortem, following removal of the cornea for transplantation.
Then they took a small sample from the macula and removed the cells to leave a thin layer of membrane which supports the photoreceptors called rod and cone cells and is where disease begins.
They analysed the proteins present in the membrane from 30 people using mass spectrometry, which identifies protein components based on their mass, to find differences in the tissue make-up between those with and without genetic risk of AMD.
The mass spectrometry, identified a series of enzymes which are made almost exclusively by mast cells. In tissue from an additional 53 people, higher levels of mast cells were found in patients with higher disease risk.
Dr Unwin added: “We next need to look at how mast cells are activated, and whether by preventing, or clearing mast cell activation we can slow or stop disease development. There are several researchers and companies looking at complement mediated-therapies for AMD and while these are promising for Chr1-related disease there is no evidence that they will have an effect on Chr10 disease. A therapy designed to target mast cell activation as a unified mechanism could in theory treat all patients with AMD and prevent sight loss.”
Researchers report in the journal Cell on insights they have made into the molecular process by which bacteria such as the highly contagious Shigella suppresses the immune system, preventing it from recognising impending infection.
Interferons are the first line of the immune system’s defence against infection. These warn neighbouring cells and prepare them to fight off an incoming infection. Many viruses – including SARS-CoV2 – have evolved proteins which inhibit normal interferon functions in order to increase infectivity.
Whether bacteria such as Shigella, which causes dysentery, are also able to interfere with the immune system and its capacity to fight the infection was previously unknown. Shigella is highly contagious, requiring only a small inoculum (10 to 200 organisms) to cause an infection.
The study found that Shigella inject a protein called “OspCs” into cells, which blocks the host’s interferon response, allowing the bacteria to successfully infect the host.
Interestingly, OspCs blocked interferon signalling by preventing cells from adapting to changing concentrations of calcium – a molecular signal that usually alerts a cell to infection and damage.
This newly identified strategy tricks the immune system, preventing the body from mounting an effective immune response to infection by decoding host calcium signals.
“This study was another perfect example of how studying pathogens can not only lead to a better understanding of infectious processes, but can also reveal the complexity of host responses to infection,” said Dr Charlotte Odendall.
These findings may be the first steps towards new bacterial treatments in the future, the researchers said.
Scanning electron micrograph of Lassa virus budding off a Vero cell. Image credit: National Institute of Allergy and Infectious Diseases, NIH
The National Institute for Communicable diseases has reported that a case of Lassa fever was diagnosed in a man from KwaZulu-Natal on 12 May 2022. The man had extensive travel history in Nigeria before returning to South Africa. He fell ill after entering South Africa and was hospitalised in a Pietermaritzburg hospital. The diagnosis of Lassa fever was confirmed by lab tests. Sadly, the man succumbed to the infection.
Contact tracing and monitoring is underway. No secondary cases of Lassa fever have been confirmed at the time of this report. In February 2022, three cases of Lassa fever had been reported in the UK, with the first travelling from Mali and the other two resulting from secondary transmission.
Originally discovered in 1969, Lassa fever is a rodentborne viral haemorrhagic fever endemic to West African countries and is caused by Lassa virus. Up to 300 000 cases of Lassa fever, with about 5000 deaths, are recorded annually in the endemic countries. Currently there is no vaccine for Lassa fever. The clinical course of Lassa fever is either not recognised or mild in 80% of patients; however, about 20% of patients might experience severe disease, including facial swelling, hepatic and renal abnormalities, pulmonary oedema, and haemorrhage. Although overall case-fatality rates for patients with Lassa fever is about 1%, rates among hospitalised case-patients are >15%. Intravenous administration of the antiviral drug ribavirin has become the standard of care for treatment of Lassa fever, but data on the efficacy of intravenous ribavirin are limited. The original study among Lassa fever patients in Sierra Leone found survival to be significantly higher (p = 0.0002) among those who obtained ribavirin within the first 6 days of illness (55%) compared with those who never received the drug (5%).
The natural reservoir of this virus in endemic countries is the Mastomys rat. The rats are persistently infected, shedding the virus in their urine and faeces. Humans can come into contact with the virus through direct contact or inhalation of the virus in areas that are infested with the infected rats. For example, contact with contaminated materials, ingestion of contaminated food or inhalation of air that has been contaminated with urine droplets. Person-to-person transmission of the virus does not occur readily and the virus is not spread through casual contact.
Person-to-person transmission is not common and is mostly associated with the hospital-setting where healthcare workers have contact with the infected blood and bodily fluids of a patient. Cases of Lassa fever in travellers returning from endemic countries are reported from time-to-time. In 2007 a case of Lassa fever was diagnosed in South Africa. That case involved a Nigerian citizen with extensive travel history in rural parts of Nigeria before falling ill, and he received medical treatment in South Africa. There were no reported secondary cases of Lassa fever on this occasion. Recently, in February 2022, an imported case of Lassa fever with secondary cases were identified in the United Kingdom.
Since the 1990s, scientists have debated the underlying cause of Gulf War illness (GWI), a constellation of unexplained and chronic symptoms affecting veterans of the Persian Gulf War. Now researchers have solved the mystery, showing through a detailed genetic study that the nerve gas sarin was largely responsible for the syndrome. The findings were published in Environmental Health Perspectives.
Dr Haley’s research group not only discovered that veterans with exposure to sarin were more likely to develop GWI, but also found that the risk was modulated by a gene that helps break down the nerve gas. Sarin-exposed Gulf War veterans with a weak variant of the gene were more likely to develop symptoms of GWI than other exposed veterans with the strong form of the gene.
“Quite simply, our findings prove that Gulf War illness was caused by sarin, which was released when we bombed Iraqi chemical weapons storage and production facilities,” said Robert Haley, MD, at UT Southwestern, a medical epidemiologist who had led that study and has been investigating GWI for 28 years. “There are still more than 100 000 Gulf War veterans who are not getting help for this illness and our hope is that these findings will accelerate the search for better treatment.”
Multiple causes of Gulf War illness suggested
After the Gulf War, more than a quarter of the US and coalition veterans began reporting a range of chronic symptoms, including fatigue, fever, night sweats, memory and concentration problems, difficulty finding words, diarrhoea, sexual dysfunction, and chronic body pain. Since then, military and academic researchers have studied a list of possible causes of GWI, ranging from stress, vaccinations, and burning oil wells to exposure to pesticides, nerve gas, anti-nerve gas medication, and depleted uranium used in weapons.
“What makes this new study a game-changer is that it links GWI with a very strong gene-environment interaction that cannot be explained away by errors in recalling the environmental exposure or other biases in the data.”
Study leader Robert Haley, MD, medical epidemiologist
Over the years, these studies have identified statistical associations with several of these, but no cause has been widely accepted. Most recently, Dr Haley and a colleague reported a large study testing veterans’ urine for depleted uranium that would still be present if it had caused GWI and found none.
Studies have shown statistical associations with several of these causes, though none received wide acceptance. Dr Haley and a colleague recently reported a large study that found no depleted uranium in veterans’ urine, which would have still been present if it had caused GWI.
“As far back as 1995, when we first defined Gulf War illness, the evidence was pointing toward nerve agent exposure, but it has taken many years to build an irrefutable case,” said Dr Haley.
Sarin’s effects
Sarin is a toxic nerve agent, production of which was banned in 1997. When people are exposed to either the liquid or gas form, sarin enters the body through the skin or breathing and attacks the nervous system. High-level sarin often results in death, but studies on survivors have revealed that lower-level sarin exposure can lead to long-term impairment of brain function. A large release of this gas occurred when a chemical weapons storage plant was bombed, causing thousands of nerve gas alarms to sound.
Previous studies have found an association between Gulf War veterans who self-reported exposure to sarin and GWI symptoms. However, this has raised criticisms of recall bias. “What makes this new study a game-changer is that it links GWI with a very strong gene-environment interaction that cannot be explained away by errors in recalling the environmental exposure or other biases in the data,” Dr Haley said.
In the new paper, Dr Haley and his colleagues studied 508 deployed veterans with GWI and 508 deployed veterans who did not develop any GWI symptoms. They asked whether the veterans had heard chemical nerve gas alarms, indicating sarin exposure, and also collected blood and DNA samples.
The role of PON1
The researchers tested the samples for variants of a gene called PON1, which has two variants. The Q variant generates a blood enzyme that efficiently breaks down sarin while the R variant helps the body break down other chemicals but is not efficient at destroying sarin. Everyone has either a QQ, RR or QR genotype.
For Gulf War veterans with the QQ genotype, hearing nerve agent alarms — a proxy for chemical exposure — raised their chance of developing GWI by 3.75 times, those with the QR genotype had an a 4.43 fold risk increase. And for those with RR genotype, the chance of GWI increased by 8.91 times. Those soldiers with both the RR genotype and low-level sarin exposure were over seven times more likely to get GWI due to the interaction per se, over and above the increase in risk from both risk factors acting alone. For genetic epidemiologists, this number leads to a high degree of confidence that sarin is a causative agent of GWI.
“Your risk is going up step by step depending on your genotype, because those genes are mediating how well your body inactivates sarin,” said Dr Haley. “It doesn’t mean you can’t get Gulf War illness if you have the QQ genotype, because even the highest-level genetic protection can be overwhelmed by higher intensity exposure.”
This kind of strong gene-environment interaction is considered a gold standard for showing that an illness like GWI was caused by a particular environmental toxic exposure, he added. The research doesn’t rule out that other chemical exposures could be responsible for a small number of cases of Gulf War illness. However, Dr. Haley and his team carried out additional genetic analyses on the new data, testing other factors that could be related, and found no other contributing causes.
“There’s no other risk factor coming anywhere close to having this level of causal evidence for Gulf War illness,” said Dr Haley.
The team is continuing research on GWI’s impacts on the body, particularly the immune system, whether any of its effects are reversible, and whether there are biomarkers to detect prior sarin exposure or GWI.
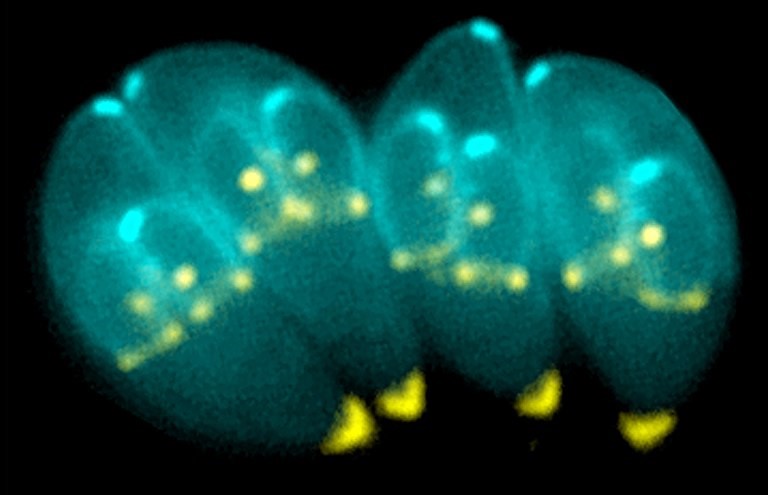
Toxoplasma gondii, an obligate intracellular human parasite, has a unique cytoskeletal apparatus that is probably used for invading host cells and for parasite replication. Shown here are images of T. gondii constructing daughter scaffolds within the mother cell. Green: YFP-α-Tubulin; bright yellow: mRFP-TgMORN1 (see Hu et al., Figure 6 A-C). Credit: Image provided by Ke Hu and John M. Murray
A new method blocks the protein regulation of Toxoplasma gondii, causing the parasite to die off inside the host cell, a method which could be adapted to malaria. The approach is detailed in the journal Nature Microbiology.
Toxoplasmosis is one of the most widespread zoonoses worldwide. It is an infectious disease that can be transmitted from cats to humans, and from consumption of raw or undercooked meat. Infection is particularly dangerous for pregnant women, and immunosuppressed HIV/AIDS patients often manifest neurological symptoms.
The cause of the disease is the single-celled parasite T. gondii. Inside the host cell, it forms a parasitophorous vacuole, a tiny compartment facilitating nutrient exchange and synchronised cell division. Up to 64 daughter cells can form inside, connected with each other inside the vacuole via a network. As soon as the offspring are mature, a regulation mechanism prompts the dissolution of the vacuole and the structures that have formed inside it, releasing the daughter cells to invade new host cells.
Hope for the development of new drugs
Previously, it was not known which genes encode the proteins that control the exit from the host cell. To identify them, a team led by Professor Markus Meißner at LMU, collaborated with colleagues from the University of Glasgow in Scotland to develop a novel genetic screening technique based on Cas9 ‘genetic scissors’, and investigate a library of 320 parasite-specific genes. They discovered two genes without which cell egress is impossible.
The targeted destruction of these genes resulted in the exit being trapped and the next generation of parasites dying within the host cell. “This paves the way potentially for the development of active substances that could block the function of the corresponding proteins and so put a halt to propagation,” remarked Prof Markus Meißner.
T. gondii is closely related to the malaria pathogen Plasmodium falciparum. Therefore, the parasite serves as a model organism for the pathogen of the tropical disease, which kills hundreds of thousands of people worldwide every year. “We assume that similar processes control the propagation of the malaria pathogen,” explains LMU parasitologist Dr. Elena Jimenez-Ruiz. “Next, we will investigate what functions these proteins have in the malaria pathogen and whether there are possible starting points for the development of new drugs.”
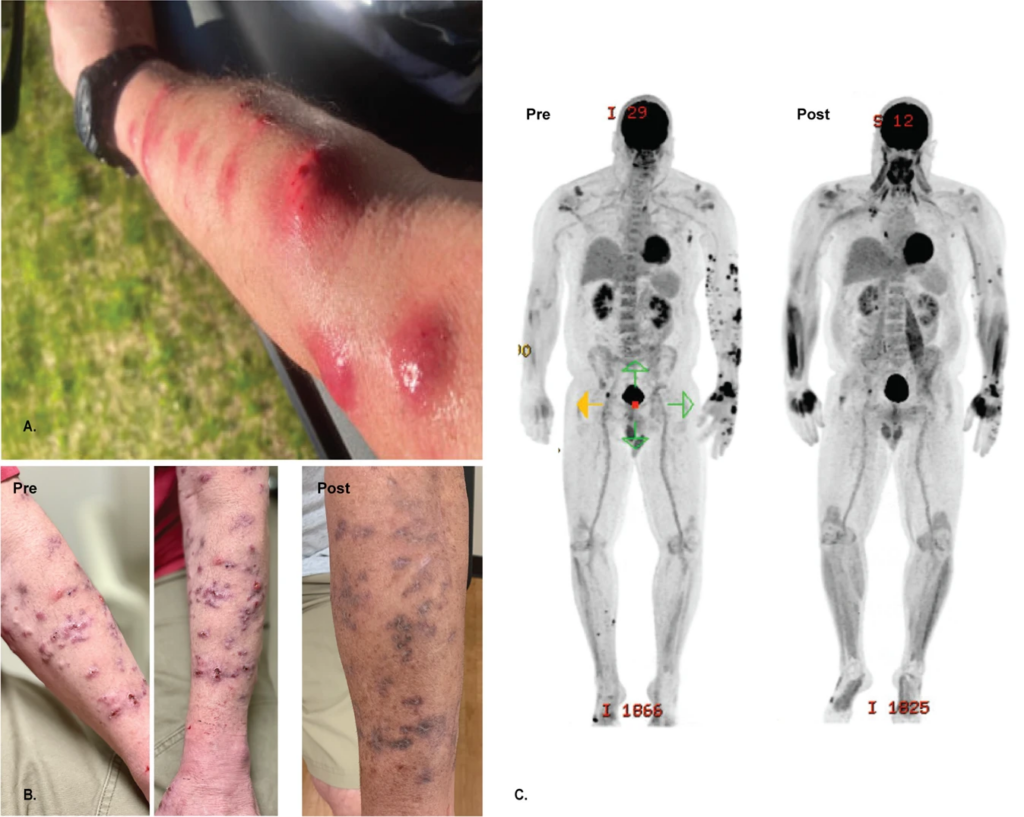
A Left upper extremity with multiple large erythematous, fluctuant to nodular lesions ultimately diagnosed as disseminated cutaneous Mycobacterium chelonae infection. B Images of the left upper extremity lesions prior to (Dec 2020) and following (August 2021) addition of bacteriophage therapy. C PET/CT prior to (March 2021) and following (August 2021) addition of bacteriophage therapy. Credit: Nature
Reporting in the journal Nature, clinicians describe the use of a bacteriophage to treat a flesh-eating infection by an antibiotic-resistant bacteria, with excellent clinical response. Bacteriophages, from the Greek ‘bacteria eater’, are viruses which target bacteria.
Bacteriophage (or more simply ‘phage’) therapy is being explored as a solution to the growing threat of antimicrobial resistance. Despite the exotic-sounding name, bacteriophage therapy is nothing new – in fact, its first application in 1919 predates the discovery of penicillin in 1929. However, their use has not been accompanied with robust research, meaning that there is still uncertainty regarding their use in modern medicine.
The authors report treating Mr. M, a 56 year-old man with disseminated cutaneous Mycobacterium chelonae infection with a single bacteriophage in conjunction with antibiotic and surgical management. He had previously received extensive antimicrobial courses as well as surgical debridement, but the bacterial infection persisted.
M. chelonae is a rapidly growing nontuberculous mycobacterium, ubiquitous in the environment and is known to have antimicrobial resistance. In rare cases, it causes infections in immunocompromised patients. To treat the infection, the researchers used a bacteriophage called Muddy, which had been isolated from a South African eggplant.
After the phage therapy skin started, lesions significantly improved both on examination and in PET/CT scans. Furthermore, two biopsies at two and five months post-treatment revealed no evidence of granulomas or AFB on histopathology and tissue cultures have remained negative. The patient has had no adverse events from the phage therapy and administered the intravenous therapy at home for more than six months.
Bacteriophage therapy is hampered by the development of phage resistance, which can potentially be countered using an appropriately-designed phage cocktail. In this case, the researchers were limited to Muddy, since no other phages tested were highly active against the patient’s strain of M. chelonae. Although resistance to Muddy is likely to occur, it was not detected in vitro, consistent with the infrequency of phage resistance in M. abscessus isolates. Resistance in vivo leading to loss of treatment efficacy was also not observed, which together suggest that phage resistance of NTM pathogens may not be the impediment encountered with other pathogens.
A second barrier to the successful treatment of bacterial infections with phage therapy is the complex interaction between the host immune system and the bacteriophage. In this case, the patient maintained stably improved disease and negative microbiologic and histopathologic studies despite a neutralising antibody response to the phage.
The authors suggested that the phage quickly reduced the burden of infection, allowing the ongoing antimicrobial therapy to have an effect. The phage also became self-replicating at the infection site – administration after the onset of neutralising antibodies therefore became unnecessary.
There are still significant challenges to phage therapy becoming widespread. The mains ones are 1) doctors need to know the bacterial strain behind the infection and 2) they need to have several phages on hand that specifically target that strain. Compounding the latter problem, most pharmaceutical companies are hesitant to focus on developing phage therapies. Since phage therapy is over 100 years old, it is difficult to patent and generate revenue to justify the initial development costs.